差异表达分析三剑客:DESeq2、edgeR、limma到底该怎么选?
但不同类型的数据在软件选择上存在差异,即便是利用便捷的在线工具,也应当关注选用不同软件可能对结果产生的不同影响。将RNA-seq计数数据转换为连续值后,使用线性模型分析。适合处理多因素实验设计(如时间序列、药物梯度),例如在分析不同浓度药物处理的转录组数据时,可直接在模型中纳入浓度作为协变量。:通过负二项分布模型和自动离散度估计:先极大似然估计,再拟合趋势线,最后修正异常值,特别擅长处理低重复样本
在RNA-seq、微阵列等基因差异表达分析中,DESeq2、edgeR和limma是最常用的三大工具。之前我们分别介绍了这三款软件,它们都可以在Galaxy云平台(网址:usegalaxy.cn)方便地使用。但不同类型的数据在软件选择上存在差异,即便是利用便捷的在线工具,也应当关注选用不同软件可能对结果产生的不同影响。但究竟该用哪一个?本文将从数据类型、样本量、算法原理到可视化方法等,带你看懂它们的核心差异。
这三款软件,由于采用了不同的统计模型和前提假设,因此各自适用于特定的数据特征和分析场景。它们在对离散度管理、低表达基因的处理等方面展现出显著的不同,可以根据以下具体情况选择:
|
情况描述 |
推荐工具 |
|
单细胞RNA-seq |
DESeq2 |
|
微阵列数据 |
limma |
|
跨组学联合分析 |
edgeR |
|
同时分析mRNA+miRNA |
limma |
|
需要复杂实验设计 |
edgeR |
|
3组生物学重复样本 |
DESeq2/edgeR |
|
样本少且质量差 |
DESeq2 |
|
样本量>7/组 |
limma |
|
大数据集(>100样本) |
limma+DESeq2组合 |
|
原始计数 |
DESeq2/edgeR |
|
分析差异剪接 |
limma |
|
关注低表达基因 |
edgeR |
-
• 注:当数据量>100样本时,先用limma快速筛选候选基因,再用DESeq2进行深度验证。
工具定位
DESeq2:小样本(侧重稳定型)
-
• 适用数据:专为RNA-seq原始计数数据设计,尤其擅长处理零膨胀数据和高变异基因。
-
• 样本量:最少3个/组(推荐≥5)
-
• 为什么用它:通过负二项分布模型和自动离散度估计:先极大似然估计,再拟合趋势线,最后修正异常值,特别擅长处理低重复样本。比如单细胞测序初步筛选差异基因时,即使只有3个重复也能稳定输出结果。
-
edgeR:灵活多变(适合精细调节)
-
• 适用数据:计数数据(支持无重复设计) 低计数基因的「狙击手」
-
• 样本量:4-6个/组最佳
-
• 为什么用它:采用精确检验和广义线性模型,在中等样本量和复杂实验设计(比如多因素交互作用)中表现亮眼。
同样基于负二项分布,但引入经验贝叶斯方法压缩离散度参数,在检测低表达差异基因时灵敏度更高。支持ChIP-seq、ATAC-seq等其他计数数据。例如研究药物剂量梯度效应时,能处理不同时间点+不同浓度的组合数据。 -
limma:大样本首选
-
• 适用数据:经过转换的标准化数据(配合
voom处理RNA-Seq) -
• 样本量:≥7个/组
-
• 为什么用它:基于线性模型和经验贝叶斯收缩,在大规模临床队列数据中计算速度碾压其他工具。limma最初为微阵列设计,通过voom转换将RNA-seq计数数据转换为连续值后,使用线性模型分析。适合处理多因素实验设计(如时间序列、药物梯度),例如在分析不同浓度药物处理的转录组数据时,可直接在模型中纳入浓度作为协变量。
-
算法核心差异
模型底层逻辑
-
• DESeq2
基因特异性离散度估计 → 负二项分布拟合趋势线 → Wald检验 压缩异常值 -
• edgeR
跨基因离散度收缩 → 广义线性模型 → 准似然F检验。通过estimateGLMTagwiseDisp函数,用全基因组信息修正单个基因离散度 -
• limma
voom权重转换 → 线性建模 → 经验贝叶斯调整。 limma的diffSplice函数可同时检测差异表达和差异剪接。
三者对比表
|
特性 |
DESeq2 |
edgeR |
limma |
|
输入要求 |
原始计数矩阵 |
原始计数矩阵 |
标准化后的表达矩阵 |
|
最小样本量 |
3个/组(推荐≥5) |
2个( 4-6个/组最佳 ) |
7(≥10个/组) |
|
优势场所 |
小样本、测序深度低 |
低计数基因、技术重复 |
大规模数据、多组比较 |
|
数据类型 |
原始数据(overdispersed count数据) |
原始计数矩阵 |
标准化后的表达矩阵 |
|
默认检验方法 |
Wald检验(负二项分布拟合趋势线) |
准似然F检验(广义线性模型 ) |
经验贝叶斯t检验(线性建模) |
|
离散度处理 |
基因间+基因内双重调整 |
基因间信息共享 |
全局经验贝叶斯收缩 |
|
多重检验校正 |
Benjamini-Hochberg |
Benjamini-Hochberg |
Bonferroni 自适应t分布调整 |
|
特色功能 |
自动过滤低表达基因 |
支持零膨胀权重 |
差异剪接分析 |
|
内存占用 |
较高 |
中等 |
较低 |
-注:limma在≥7样本/组时,其经验贝叶斯方差收缩算法能显著提升统计效能。
可视化对比
-
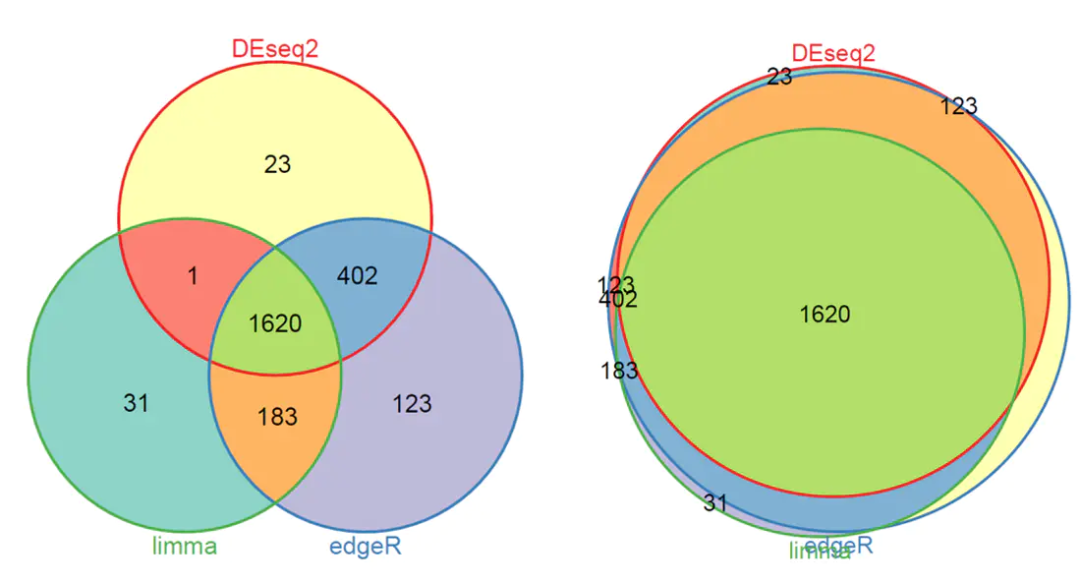
1. 韦恩图:工具重叠比较(三工具差异基因交集 )
-
• DESeq2特有基因:与细胞周期相关
-
• limma特有基因:涉及代谢通路
-
• 三者共有基因:经典癌症标记物(如TP53)
-
-
2. 火山图:各工具显著性分布
-
-
• DESeq2:显著性基因更集中
-
• edgeR:检测到更多低表达差异基因
-
• limma:分布最对称
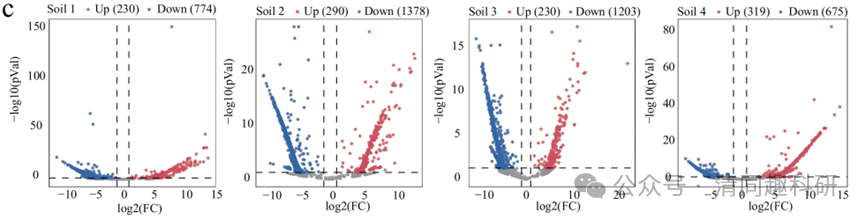
-
3. 热图:top50差异基因表达模式 (limma差异基因聚类)
-
• DESeq2结果:聚类结构最清晰
-
• limma结果:包含更多弱效应基因。要是样本量超过60个,limma的线性模型优势就开始显现
-
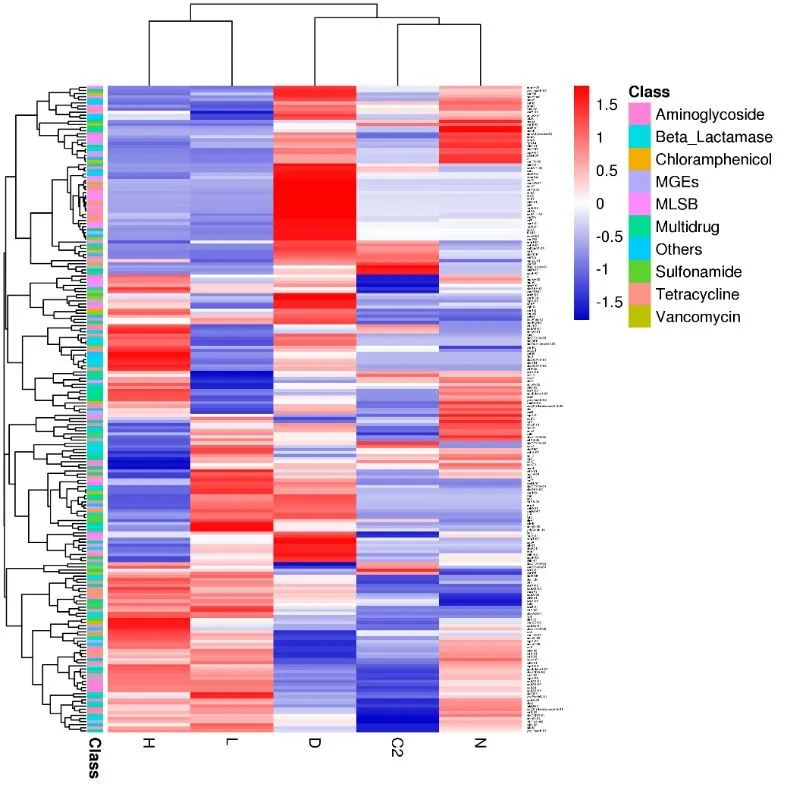
-
-
推荐阅读
中国银河生信云平台(UseGalaxy.cn)以“让生信分析更简单”为使命。平台致力于为科研工作者、医疗机构和生物产业技术人员提供全栈式生物信息学分析解决方案。
优先技术响应、定制化工具部署、阶梯式能力培养,请加入「Galaxy生信星球」。咨询微信:usegalaxy 或 galaxy-help


开源鸿蒙跨平台开发社区汇聚开发者与厂商,共建“一次开发,多端部署”的开源生态,致力于降低跨端开发门槛,推动万物智联创新。
更多推荐
 13
13 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)