肿瘤“内鬼”现形记:inferCNV一眼锁定恶性单细胞
inferCNV用于分析肿瘤单细胞RNA测序数据,通过比较肿瘤细胞与参考"正常"细胞的基因表达强度,识别体细胞大规模染色体拷贝数变异的证据(如整条染色体或大片段染色体的扩增或缺失)。该方法可生成展示肿瘤基因组各染色体区域相对表达强度的热图,相较于正常细胞,肿瘤基因组中过度富集或缺失的区域通常会直观显现。换一种说法就是“inferCNV把肿瘤单细胞基因组当成‘唱片’——让正常参考细胞当‘原唱’,肿瘤
简介:
inferCNV用于分析肿瘤单细胞RNA测序数据,通过比较肿瘤细胞与参考"正常"细胞的基因表达强度,识别体细胞大规模染色体拷贝数变异的证据(如整条染色体或大片段染色体的扩增或缺失)。该方法可生成展示肿瘤基因组各染色体区域相对表达强度的热图,相较于正常细胞,肿瘤基因组中过度富集或缺失的区域通常会直观显现。换一种说法就是“inferCNV把肿瘤单细胞基因组当成‘唱片’——让正常参考细胞当‘原唱’,肿瘤上皮细胞当‘翻唱’;哪段旋律(染色体区域)突然飙高或静音,热图里立刻亮起红灯,恶性细胞的‘跑调’证据瞬间被抓包。”
注意:参考细胞一般选择正常的细胞,比如这里的肿瘤样本中的上皮细胞是观察组,我们可以选择正常样本中的上皮细胞作为参考,如果没有正常样本,可以选择肿瘤样本中的免疫细胞。
环境配置:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c('Seurat', 'rjags', 'infercnv','qs','AnnoProbe','ggplot2', 'dplyr', 'gplots'))数据准备:
通过网盘分享的文件:inferCNV
链接: https://pan.baidu.com/s/1CpdXQCCcjCX8m_cPFMXgfw 提取码: 6vqs
分析流程:
#加载R包和数据
rm(list = ls())
suppressMessages({
library(rjags)
library(infercnv)
library(Seurat)
library(gplots)
library(ggplot2)
library(qs)
library(AnnoProbe)
})
setwd("/share/nas1/sunh/project/zhihu/06_inferCNV/")
sce <- readRDS('/share/nas1/sunh/project/zhihu/06_inferCNV/test.rds')查看数据细胞分类
table(sce$celltype) #该示例数据提取了部分数据,其中0-6群是上皮细胞大类进行亚分群得到的,B细胞用于做参考细胞
数据预处理,创建InferCNV对象前需要准备好三个文件:conut matrix;cell type annotations;gene ordering file
# 文件1:表达量矩阵
# 除了用GetAssayData函数其实也可以直接sce@assays$RNA@count即可
dat <- GetAssayData(sce,layer = 'counts',assay = 'RNA')
# 文件2:样本的描述
groupinfo <- data.frame(v1 = colnames(dat),v2 = sce@meta.data$celltype)
head(groupinfo)
# 文件3:基因在染色体中的坐标
#注释信息可能包括基因名称、染色体位置、基因描述等。
geneInfor <- annoGene(rownames(dat),"SYMBOL","human") #物种需要小心哦
# 移除包含 chrM, chrX, 和 chrY 的行
geneInfor <- geneInfor[!geneInfor$chr %in% c("chrM", "chrX", "chrY"), ]
#sub函数用于把chr替换为空
geneInfor$chr_num <- as.numeric(sub("chr", "", geneInfor$chr))
colnames(geneInfor)
#with函数的作用是简化写法,可问gpt
geneInfor <- geneInfor[with(geneInfor,order(chr_num,start)),c(1,4:6)]
geneInfor <- geneInfor[!duplicated(geneInfor[,1]),]
head(geneInfor)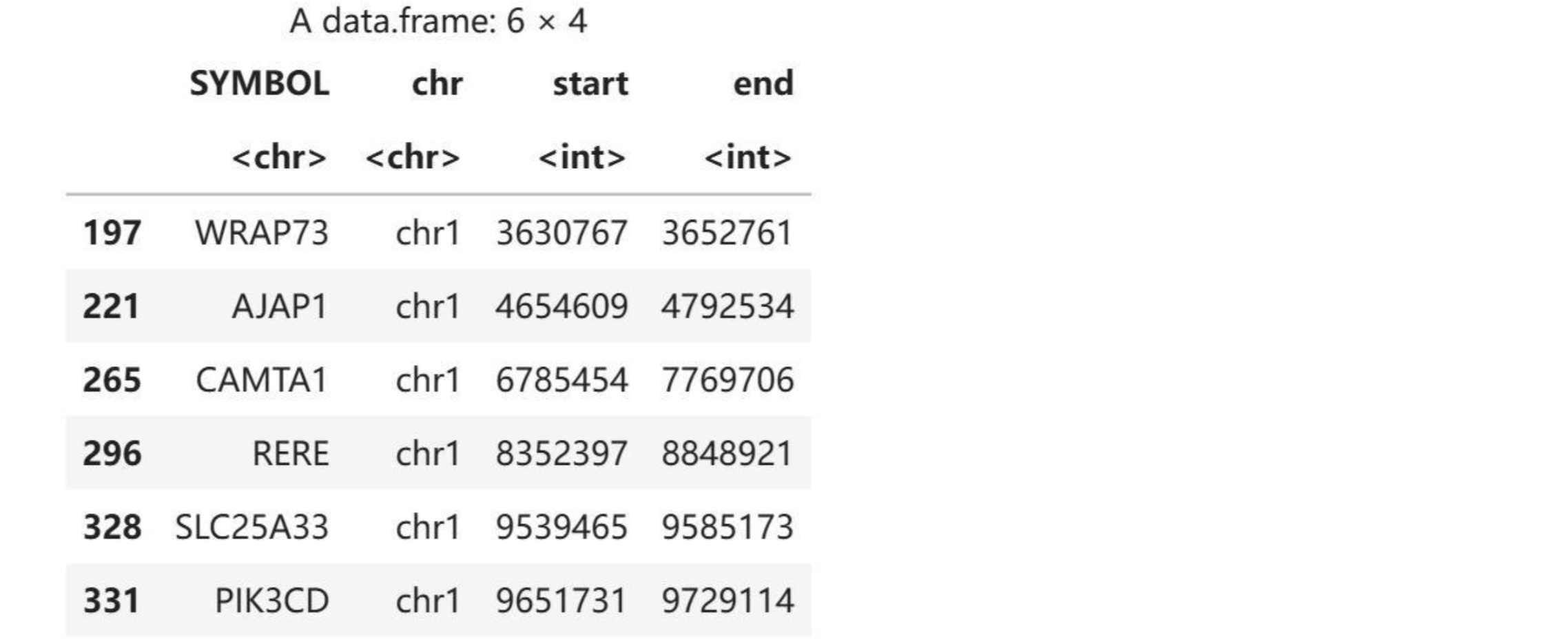
#保留行名在geneInfor第一列中存在的行。
dat <- dat[rownames(dat) %in% geneInfor[,1],]
#获得位置后对dat进行重新排序使其跟geneInfor中的顺序一致
dat <- dat[match(geneInfor[,1],rownames(dat)),]
# 保存矩阵文件
#expFile:是一个变量,存储写入的文件的文件名或路径。在这里文件名是expFile.txt。
expFile <- 'expFile.txt' #定义输出文件名
colnames(dat) <- gsub("-", "_", colnames(dat))
write.table(dat, file = expFile, sep = '\t', quote = F)
# 保存细胞注释文件
groupFiles <- 'groupFiles.txt'
groupinfo$v1 <- gsub("-", "_", groupinfo$v1)
write.table(groupinfo,file = groupFiles, sep = '\t', quote = F, col.names = F, row.names = F)
# 保存基因排序文件
head(geneInfor)
geneFile <- 'geneFile.txt'
write.table(geneInfor, file = geneFile, sep = '\t',quote = F, col.names = F, row.names = F)创建infercnv对象
infercnv_obj <- CreateInfercnvObject(raw_counts_matrix = expFile,
annotations_file = groupFiles,
delim = "\t",
gene_order_file = geneFile,
ref_group_names = c("B cell") #这里用B cell做参照
)
infercnv_obj <- infercnv::run(infercnv_obj,
cutoff = 0.1, #smart-seq选择1,10X选择0.1
out_dir = "infercnv_output", # 命名输出文件夹名称
cluster_by_groups = TRUE, #是否根据细胞类型(由细胞注释文件中定义)
hclust_method = "ward.D2",# ward.D2 方法进行层次聚类
analysis_mode = "subclusters", # 默认是samples,推荐是subclusters
denoise = TRUE, # 去噪音
HMM = F, ##特别耗时间,是否要去背景噪音
plot_steps = F, #不在每个步骤后生成图形。
leiden_resolution = "auto", #可以手动调参数
num_threads = 15 # 15线程工作,加快速度,一般电脑不建议哈
)
infercnv_obj_medianfiltered = infercnv::apply_median_filtering(infercnv_obj)
infercnv::plot_cnv(infercnv_obj_medianfiltered,
out_dir='./',
output_filename='infercnv.median_filtered',
x.range="auto",
x.center=1,
title = "infercnv",
color_safe_pal = FALSE)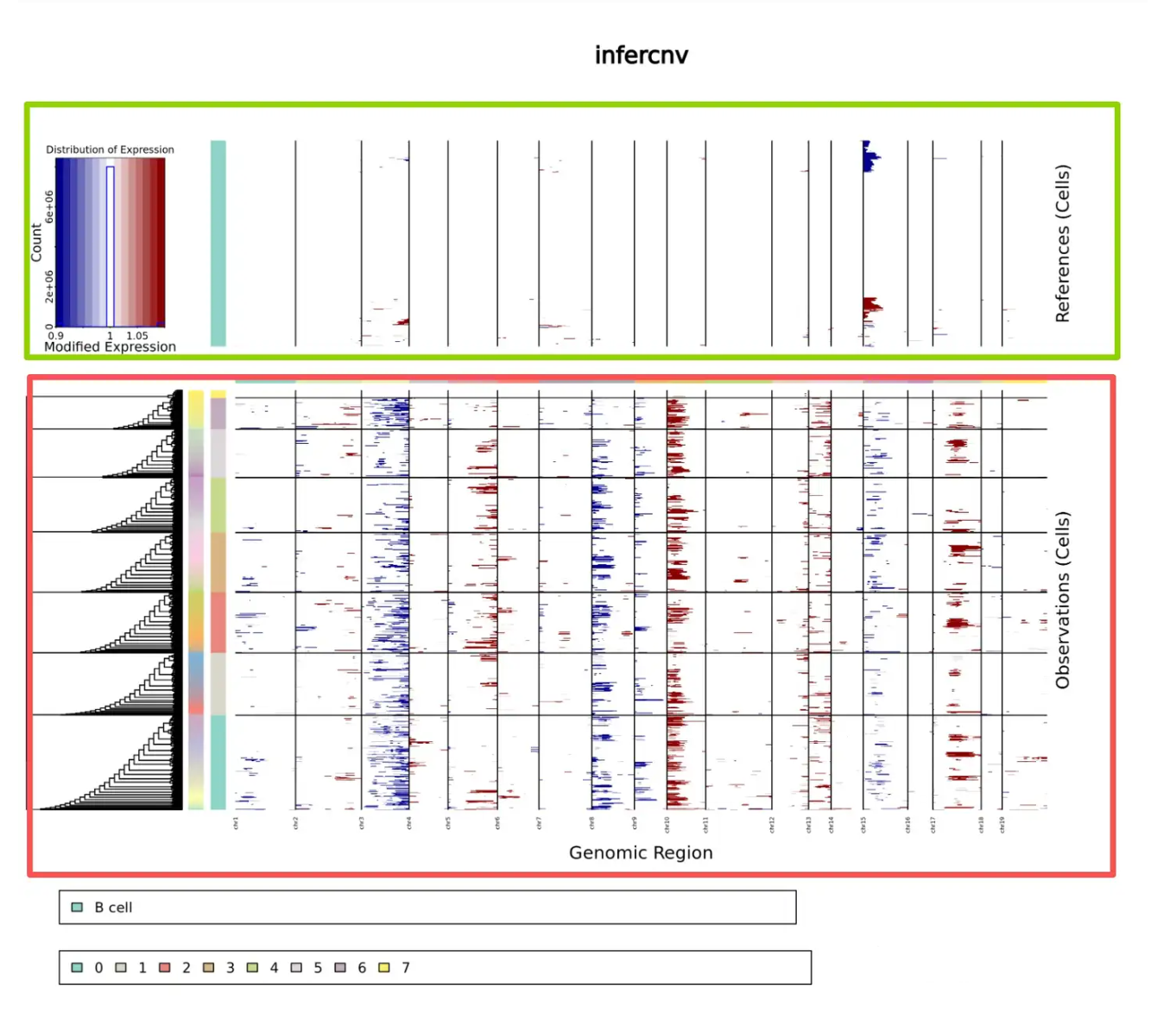

开源鸿蒙跨平台开发社区汇聚开发者与厂商,共建“一次开发,多端部署”的开源生态,致力于降低跨端开发门槛,推动万物智联创新。
更多推荐
 9
9 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)