利用序列和结构的多模态对比学习改进ncRNA家族预测
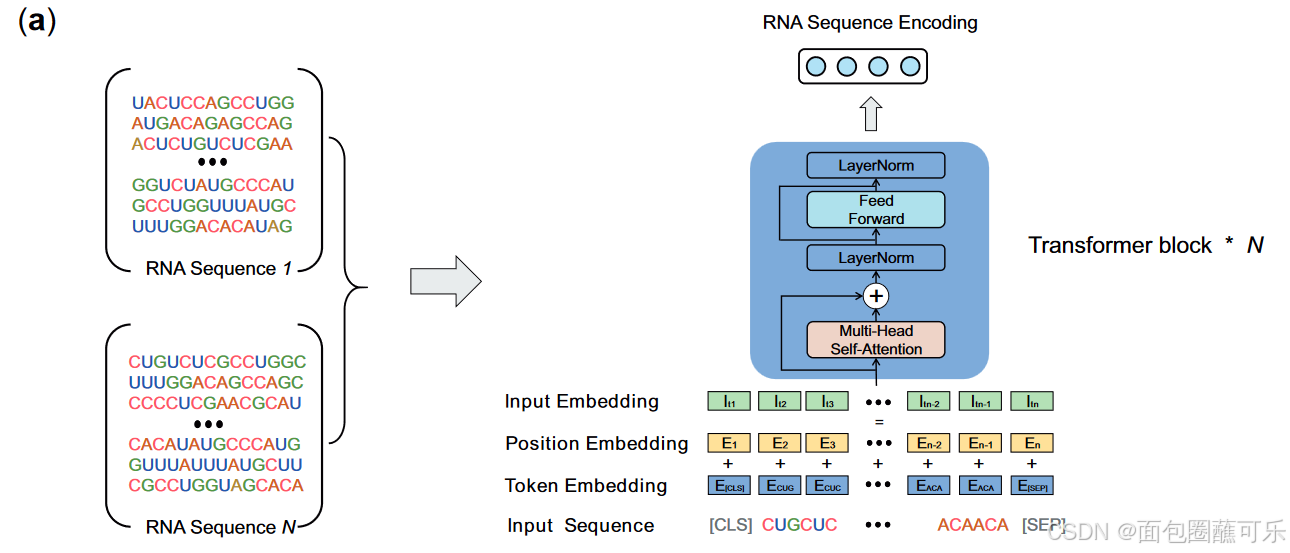
本文提出的非编码RNA家族预测模型由ncRNA序列表示学习、ncRNA结构表示学习和ncRNA家族预测3个模块组成。首先,在包含190 000个一级序列结构的大规模哺乳动物nc RNA数据集上预训练BERT。然后,我们使用图神经网络对ncRNA的二级结构进行建模,并通过对比学习的方式将其与BERT模型相结合,使得两种模型提取的特征之间能够进行交互。最后,使用注意力机制将这两类特征进行融合,然后输入
材料与方法
本文提出的非编码RNA家族预测模型由ncRNA序列表示学习、ncRNA结构表示学习和ncRNA家族预测3个模块组成。首先,在包含190 000个一级序列结构的大规模哺乳动物nc RNA数据集上预训练BERT。然后,我们使用图神经网络对ncRNA的二级结构进行建模,并通过对比学习的方式将其与BERT模型相结合,使得两种模型提取的特征之间能够进行交互。最后,使用注意力机制将这两类特征进行融合,然后输入到ncRNA家族预测的最终任务中。
非编码RNA序列表示学习
在该公式中,我们将ncRNA序列视为文本,在预训练阶段使用BERT进行编码,并使用掩码语言模型。使用k - mer方法对ncRNA序列进行预处理,其中每个k - mer (长度为k的子序列)表示为一个词。词汇是基于所有可能的k - mers构建的。对特定比例的令牌进行随机掩码,并用唯一的[掩码]令牌进行替换,从而模拟预训练时对令牌进行掩码和预测的过程。在模型中使用多传感头注意力机制来提取上下文信息:

其中WQi,WKi,WVi和WO是参数矩阵. Q、K和V分别代表查询、键和值。自注意力机制使BERT能够在其预训练阶段封装非编码RNA序列的核心语义和句法特征。我们使用来自RNACentral的190 000条ncRNA初级序列作为模型预训练的数据集。然后,我们将模型微调到如图1a所示的家庭预测任务上。
非编码RNA结构表示学习
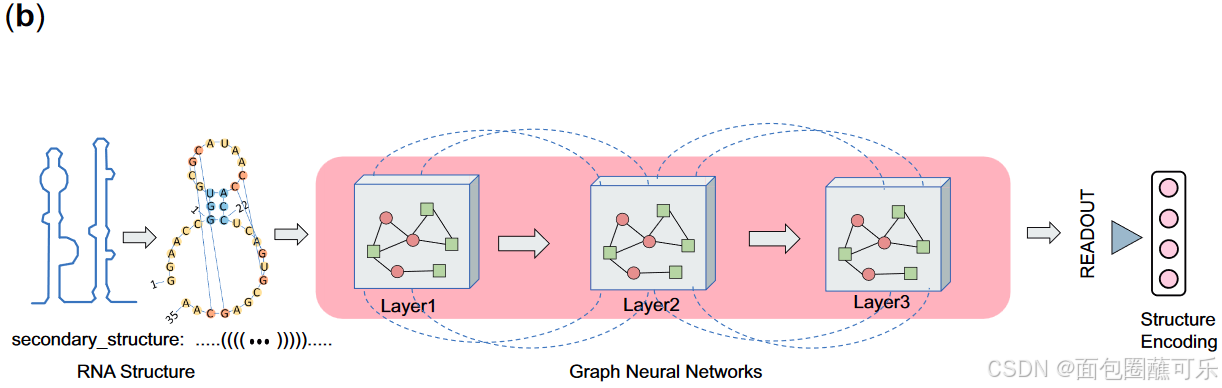
除了序列结构外,我们还加入了对ncRNA二级结构的建模。非编码RNA ( non-coding RNA,ncRNA )序列的二级结构为其拓扑结构和碱基配对相互作用提供了有价值的见解。非编码RNA ( non-coding RNA,ncRNA )的二级结构可以用图形的形式表示,其中顶点表示单个核苷酸,边表示它们之间的相互作用。图卷积网络( Graph Convolutional Networks,GCNs )作为破译nc RNA序列的拓扑属性和碱基配对的有力工具而备受关注。GCNs善于封装核苷酸的局部上下文,并在整个序列中传播信息,这对于理解RNA的二级结构至关重要。GCNs通过特征嵌入和邻域聚合,可以捕获RNA二级结构所特有的模式。这些网络的多层卷积结构有利于同时学习局部和全局上下文,从而提高模型对ncRNA分子3D结构的预测精度。
在这种情况下,一个ncRNA可以表示成一个图G =(V,E),其中V表示ncRNA分子中代表核苷酸的节点集合,E表示这些节点之间象征各种相互作用的边集合。ncRNA图中的每个核苷酸节点u链接到一个特征向量,该特征向量包含当前和邻近的核苷酸类别、是否属于形成假结的节点以及一系列物理或化学特征等相关信息。ncRNA图谱能够保留细粒度的信息,这些信息对于理解ncRNA分子的结构和功能至关重要。ncRNA序列的边连接了ncRNA分子内的相邻节点,有效地表示了核苷酸的顺序,并捕获了ncRNA分子一级结构的线性排列。我们还建立了结构边来表示形成假结的边。这些结构边的组成来源于点括号符号的规则。
对于图中的每个核苷酸ui,我们初始化其表示为。然后,我们使用k层GNN逐层计算并更新每个节点表示如下:更新后的第k+1层节点ui的隐藏状态
计算如下:其中σ是激活函数,N(ui)是ui的邻居节点的集合,cij是边eij的权重,Wk是k层的权重矩阵。这里,N(ui)是节点ui的邻居节点集合,cij是节点ui和其他邻居节点uj之间的归一化稀疏度。Wk为第k层的可学习权重矩阵,
为激活函数。计算每个节点表示的最终图层后,通过读出操作得到第n个ncRNA的图级表示Gn (图1b ),该图级表示捕获了ncRNA的结构信息。
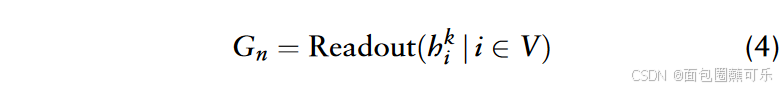
其中,Readout (·)表示一个操作,将ncRNA图中每个节点的表示转换为一个综合的d维表示,从而捕获ncRNA图的整体结构。
非编码RNA家族预测
一个给定的非编码RNA ( non-coding RNA,ncRNA )应该在其序列和结构之间表现出显著的相似性。因此,在我们设计的框架中,通过使用对比学习框架来增强相同ncRNA的序列和二级结构之间的相关性。
多模态对比学习
尤其是对比学习是ncRNA两种不同模态(序列和结构特征)之间的桥梁。它通过最小化相同ncRNA之间不同模态的距离和最大化不同ncRNA之间的距离来改进特征融合:
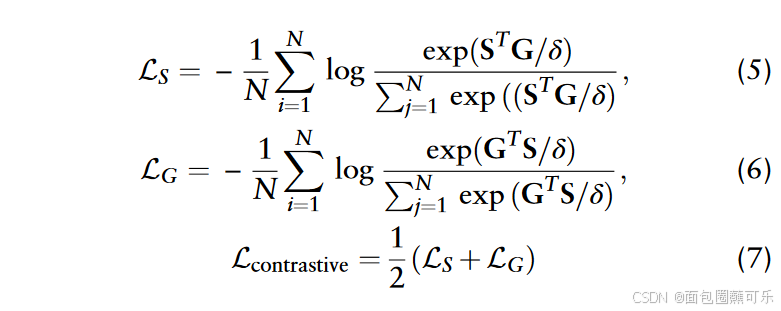
其中δ是温度参数,是一个超参数。
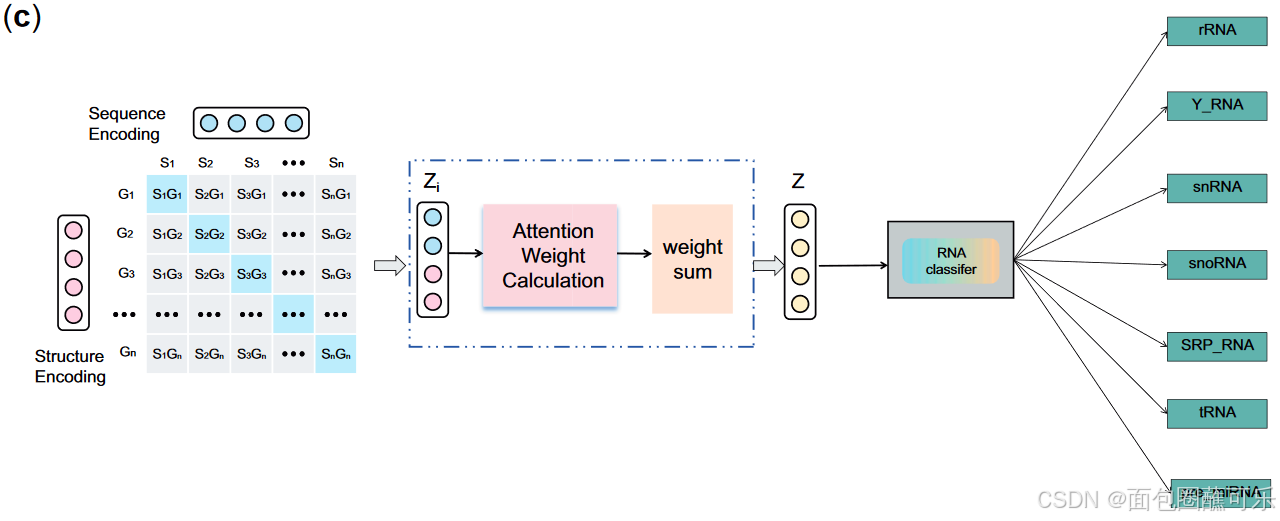
在将对比学习应用于序列和结构表征之后,我们使用注意力机制来融合这两种表征形式。
最初,序列特征和结构特征都被投影到一个共享的潜在空间中。
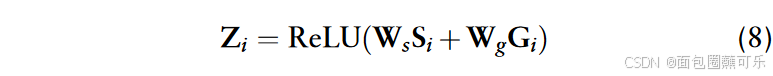
其中Si和Gi分别表示ncRNA的序列特征和结构特征,Ws和Wg是权重矩阵。
最后,利用自注意力机制确定每个特征维度在融合过程中的贡献。注意力权重使用以下公式计算:
αi的值计算如下:
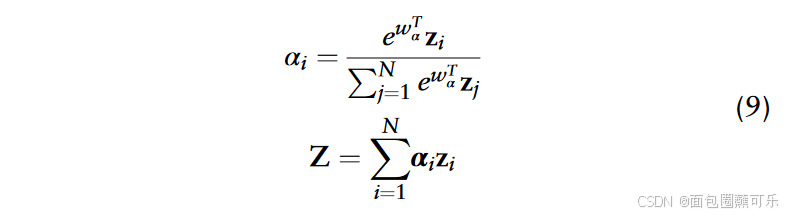
式( 9 )表示项的加总,其中每一项都是alpha系数与zi值的乘积。变量N表示求和的总项数,Z表示ncRNA的最终表示,wα为注意力权重参数, ð为特征维度数,αi为第i个特征维度的注意力权重。
家系预测
将序列和结构的融合表示用于家系预测。我们将家系预测视为多类分类任务,并据此训练多层感知机模型。为了进一步整合两种模态学习到的特征,我们设计了一个统一的损失函数,将对比学习的损失与分类任务的损失相结合,可以帮助模型更好地学习序列和结构特征之间的相关性。
首先,分类任务的损失是交叉熵损失:
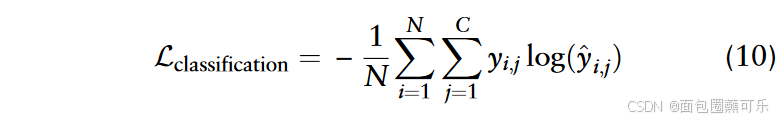
式中:是第i个样本对应于第j类的标签;
是第i个样本属于第j类的模型预测概率。损失函数将模型预测的概率分布与真实的标签分布进行比较。
模型的最终损失是两个损失的加权和:

其中,(α+β=1),α和β为超参数,调节对比学习损失的贡献及其对整个模型的贡献(图1c )。
几种数据库
Rfam是一个专门用于存储、注释和分析非编码RNA ( non-coding RNA,ncRNA )序列的数据库。这些序列包含多种ncRNA家族和结构化的RNA元件,包括RNA结构域和单位。EMBL的RNA组开发并维护了RFAM,它提供了注释、搜索和分析RNA序列和结构的工具。这一资源对于促进我们对RNA生物学和功能研究的理解具有重要意义。
GeneCards是一个在线基因数据库,为研究人员和医学专业人员提供了广泛的遗传信息。它整合了多种基因组数据,包括基因名称、别名、位置、功能、表达模式、相关疾病、通路和蛋白质相互作用等。GeneCards为基因相关研究和临床应用提供了一个全面的、用户友好的平台。
ENA (欧洲核苷酸资料库)是由欧洲生物信息学研究所管理的一个重要的核苷酸序列数据库。它与Gen Bank和DDBJ一起,在全球范围内收集、存档和传播包括DNA和RNA序列在内的核苷酸序列数据。
RNACentral是一个全面的数据库,用于存储和检索全球范围内的ncRNA序列。作为一种集中式的资源,RNACentral包含了具有记录的二级结构信息的RNA序列。当这些信息可用时,它为当前的二级结构数据提供了相关的点括号符号。
基线模型
PlncRNA-HDeep ( Meng et al 2021)使用k - mer核苷酸和one - hot编码对样本序列进行编码,便于单独训练长ncRNALSTM和CNN模型,并在决策层进行组合。使用RNA序列作为输入,PlncRNAHDeep捕获了多样化的信息,并利用了长ncRNALSTM和CNN的优势。
ncRFP ( Wang et al.2021a)是一种端到端的ncRNA家族预测方法。它仅仅依赖于序列来进行预测任务。该模型包括BiLSTM、注意力机制和全连接网络。BiLSTM和注意力机制主要负责将各种ncRNA编码成标准化的数据格式,而全连接层则用于解码输入以进行分类。
ncDLRES ( Wang et al 2021b , c )是一种基于序列特征学习的ncRNA家族预测方法,使用LSTM和ResNet。与同源序列比对方法相比,降低了对数据的要求,拓宽了适用范围。
ncDENSE ( Chen et al.2023b)使用深度学习模型,通过从ncRNA序列中提取特征来预测ncRNA家族。它使用独热编码对序列中的碱基进行编码,然后将其输入到包含Bi_GRU、DenseNet和注意力机制的集成深度学习模型中。Bi_GRU提取不同权重的特征,注意力集中在权重较高的信息上。
MFPred主要通过从ncRNA序列中提取特征来识别ncRNA家族。该模型由3个主要模块组成。第一个模块使用四种序列编码方式对序列进行特征提取和融合。第二个模块采用Bi - GRU和特征融合模块。第三个模块使用ResNetSE模块提取局部特征。本研究的主要目标是通过整合多模态信息来预测nc RNA家族。

开源鸿蒙跨平台开发社区汇聚开发者与厂商,共建“一次开发,多端部署”的开源生态,致力于降低跨端开发门槛,推动万物智联创新。
更多推荐

 19
19 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)